
Choroba, ktorej výskyt je spojený s nedostatkami genetického bunkového aparátu, fenylketonúrie, je zahrnutá v niekoľkých zoznamoch dedičných ochorení liečiteľných. Priekopníkom tohto ochorenia bol lekár z Nórska I.A. Kácením sa neskôr zistilo, že za vývoj a priebeh ochorenia je zodpovedný jeden gén nazývaný gén fenylalanín-hydroxylázy (dlhé rameno 12. chromozómu, ktorý obsahuje až 4, 5% materiálu z celkovej bunkovej DNA). Vrodená porucha vedie k čiastočnej alebo úplnej deaktivácii pečeňového enzýmu fenylalanín-4-hydroxylázy.
Ako choroba fenylketonúria
Dedičná choroba fenylketonúria (PKU) vedie k chronickej otrave tela toxickými látkami, ktoré vznikajú v dôsledku zhoršeného metabolizmu aminokyselín a procesu hydroxylácie fenylalanínu. Konštantná intoxikácia spôsobuje poškodenie centrálneho nervového systému (CNS), ktorého prejavom je progresívny pokles inteligencie (fenylpyruvická oligofrénia).
Fellingova choroba sa prejavuje nadmerným hromadením fenylalanínu v tele a jeho nesprávnym metabolizmom. Medzi ďalšie faktory vo vývoji fenylketonúrie patrí narušený transport aminokyselín cez hematoencefalickú bariéru, nízke hladiny neurotransmiterov (serotonín, histamín, dopamín). Pri absencii včasnej liečby vedie choroba k mentálnej retardácii a môže spôsobiť smrť dieťaťa.

Mechanizmus rozvoja chorôb
Kauzálnym faktorom pri výskyte génových porúch je metabolický blok, ktorý zabraňuje tvorbe fenylalanín-4-hydroxylázy (enzýmu zodpovedného za konverziu aminokyseliny fenylalanínu na tyrozín). Proteinogénna aminokyselina tyrozín je zložkou proteínov a melanínového pigmentu, preto je nevyhnutným prvkom pre fungovanie všetkých telesných systémov a jej nedostatok vedie k fermentopatii.
Dôsledkom potlačenia tvorby metabolitu spôsobeného mutačnou inaktiváciou enzýmu je aktivácia pomocných ciest na výmenu fenylalanínu. Aromatická alfa-aminokyselina v dôsledku chybných metabolických procesov sa rozkladá na toxické deriváty, ktoré sa za normálnych podmienok nevytvárajú:
- Fenylpyruvová kyselina (fenylpyruvát) je mastná aromatická alfa-keto kyselina, jej tvorba vedie k myelinizácii neuronálnych procesov a demencie;
- Kyselina fenyl-mliečna - produkt vznikajúci počas redukcie kyseliny fenylpyrohroznovej;
- Fenyletylamín - východisková zlúčenina pre biologicky aktívne vysielače elektrochemických pulzov, zvyšuje koncentráciu dopamínu, adrenalínu a noradrenalínu;
- ortofenylacetát je toxická látka, ktorá spôsobuje narušenie metabolických procesov tukových látok v mozgu.
Lekárske štatistiky naznačujú, že patologicky zmenený gén je prítomný v 2% populácie, ale neprejavuje sa. Genetický defekt sa prenáša na dieťa od rodičov len vtedy, ak je choroba prítomná u oboch partnerov a v 50% prípadov sa dieťa stáva nositeľom mutovaného génu, ktorý zostane zdravý. Pravdepodobnosť, že fenylketonúria u novorodencov povedie k ochoreniu, je 25%.
Aký typ je zdedený
Felling disease je genetická porucha dedená autozomálne recesívnym spôsobom. Tento typ dedičnosti znamená, že k vývoju príznakov vrodenej choroby dôjde len vtedy, keď dieťa zdedí jednu defektnú genoskopiu od oboch rodičov, ktorí sú heterozygotnými nosičmi modifikovaného génu.
Vývoj vrodenej choroby v 99% prípadov spôsobuje mutáciu génu zodpovedného za kódovanie enzýmu, ktorý zabezpečuje syntézu fenylalanín-4-hydroxylázy (klasická fenylketonúria). Až 1% genetických ochorení je spojených s mutačnými zmenami, ktoré sa vyskytujú v iných génoch a spôsobujú nedostatok dihydropteridín reduktázy (PKU typu II) alebo tetrahydrobiopterínu (PKU typu III).
Fenylketonúria u detí
Klasická forma genetického ochorenia u detí sa vo väčšine prípadov prejavuje v externe rozpoznateľných znakoch, počnúc 3-9 mesiacmi života. Novorodenci s chybným génom vyzerajú zdravo, charakteristickým znakom je špecifický habitus (vzhľad) dieťaťa. Ťažké príznaky sa objavujú 6-12 mesiacov po narodení.
PKU typ II sa vyznačuje tým, že prvé klinické príznaky sa objavujú po 1, 5 roku od ich narodenia. Symptómy ochorenia nezmiznú po diagnostike genetických abnormalít a začiatku diétnej terapie. Tento typ vrodenej choroby často vedie k smrti v 2-3 rokoch života dieťaťa. Najčastejšie príznaky PKU typu II sú:
- výrazné odchýlky v duševnom vývoji;
- hyperreflexia;
- porušenie motorických funkcií všetkých končatín;
- syndróm nekontrolovaných svalových kontrakcií.
- vysoký stupeň mentálnej retardácie;
- zreteľne znížená veľkosť lebky vo vzťahu k iným častiam tela;
- svalová spasticita (s možnou úplnou nehybnosťou končatín).

Prejavy kácenia choroby
V priebehu klinických štúdií a pozorovaní sa predpokladalo, že účinky toxických derivátov metabolizmu fenylalanínu spôsobujú pokles intelektuálnych schopností, ktoré sú progresívne a môžu viesť k demencii (oligofrénia, idiocia). Medzi údajnými príčinami ireverzibilných porúch mozgovej aktivity je najvhodnejší nedostatok neurotransmiterov prenášajúcich impulzy medzi neurónmi spôsobenými poklesom hladiny tyrozínu.
Presná kauzálna súvislosť medzi dedičným ochorením a mozgovými poruchami ešte nebola identifikovaná, ani vývojový mechanizmus v dôsledku fenylketonúrie takých duševných stavov, ako je ekpraxia, echolalia, záchvaty hnevu a podráždenosť. Údaje z výsledkov testov ukazujú, že fenylalanín má priamy toxický účinok na mozog, čo môže tiež spôsobiť zníženie inteligencie.
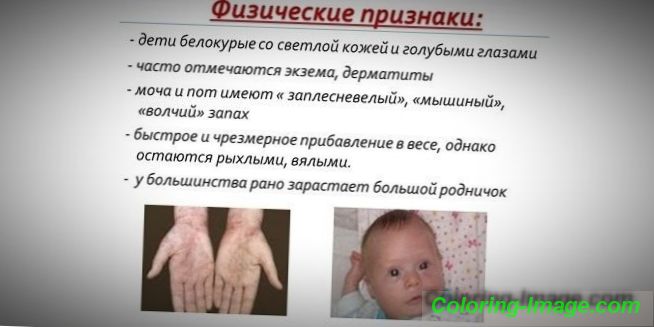
Fyzikálne a fenotypové znaky
Vzhľadom k tomu, že pigment kože a vlasov závisí od hladiny tyrozínu v mitochondriách hepatocytov a fenylketonúria zastavuje konverziu fenylalanínu, pacienti s týmto ochorením majú fenotypové znaky (recesívne symptómy). Zvýšený svalový tonus spôsobuje odchýlku v tele - stáva sa dysplastickým. Výrazné vonkajšie znaky fenylketonúrie zahŕňajú:
- hypopigmentácia - svetlá koža, svetlomodré oči, bielené vlasy;
- modravosť končatín;
- znížená veľkosť hlavy;
- špecifická poloha tela - keď sa snažíte stáť alebo sedieť, dieťa preberá postoj „krajčíra“ (ruky a nohy ohnuté v kĺboch).
Príznaky ochorenia
S včasnou detekciou je Fellingova choroba prístupná úspešnej liečbe úpravou výživy a vývoj dieťaťa je v súlade s jeho vekovou skupinou. Problémom pri detekcii mutácie génu je, že je ťažké zistiť skoré príznaky aj pre skúseného pediatra. Závažnosť symptómov vrodeného ochorenia sa zvyšuje s rastúcim dieťaťom, pretože používanie proteínových potravín prispieva k rozvoju porúch CNS.
Známky u novorodencov
Počas prvých dní života dieťaťa je ťažké zistiť príznaky patologických abnormalít - dieťa sa správa prirodzene a nie je oneskorenie vývoja. Prvé príznaky ochorenia sa začínajú objavovať v 2-6 mesiacoch po narodení. Rodičia by mali byť upozornení na správanie dieťaťa, ktoré sa vyznačuje nízkou aktivitou, letargiou alebo naopak úzkosťou, hyper vzrušivosťou.
S nástupom dojčenia, bielkoviny začnú prúdiť do tela novorodenca s mliekom, ktoré slúži ako katalyzátor pre objavenie sa prvých príznakov, ktoré jasne naznačujú, že choroba začala napredovať. Špecifické klinické prejavy ochorenia zahŕňajú: \ t
- pretrvávajúce zvracanie (často mylne považované za vrodené zúženie pyloru);
- častá regurgitácia;
- nedostatočná reakcia na vonkajšie podnety;
- svalová dystónia (znížené svalové napätie);
- kŕčovitý syndróm (záchvaty epileptického alebo neepileptického charakteru).
Symptómy u detí po 6 mesiacoch
Ak sa prejav genetického ochorenia nevyskytol (alebo nebol zaznamenaný) počas prvých šiestich mesiacov od momentu narodenia dieťaťa, potom po tomto období je už možné presne určiť psychomotorické oneskorenie. Symptómy genetických porúch spôsobených nedostatkom enzýmov u detí starších ako šesť mesiacov sú: \ t
- pokles aktivity (až do úplnej ľahostajnosti);
- žiadne pokusy postaviť sa na vlastnú päsť, sedieť;
- špeciálny "pach" kože na koži (zápach plesne vzniká v dôsledku vylučovania toxických derivátov fenylalanínu cez potné žľazy a moč);
- strata schopnosti vizuálneho rozpoznávania rodičov;
- odlupovanie kože;
- výskyt dermatitídy, ekzému, sklerodermie.
Progresia ochorenia v neprítomnosti liečby v detstve
Ak sa vývojové abnormality nezistili v detstve a primeraná liečba sa nevykonala, potom sa ochorenie začne aktívne rozvíjať a často vedie k invalidite. Nedostatok liečby v počiatočnom štádiu ochorenia spôsobuje vznik nasledujúcich príznakov ochorenia už vo veku 1, 5 roka:
- mikrocefalia (znížená veľkosť mozgu);
- prognathia (posunutie hornej chrupu dopredu);
- neskoré zuby;
- hypoplaziu skloviny (riedenie alebo úplnú neprítomnosť zubnej skloviny);
- oneskorený vývoj reči až do úplného nedostatku reči;
- 3, 4 stupeň oligofrénie (mentálna retardácia, mentálna retardácia);
- vrodené srdcové chyby (defekty v štruktúre srdcového svalu, časti srdca, veľké cievy);
- poruchy vegetatívneho systému (akrocyanóza, nadmerné potenie, arteriálna hypotenzia);
- zápcha.
Príčiny a provokujúce faktory
Pri mutáciách s autozomálne recesívnym vzorom dedičnosti musí byť defektný gén zdedený po oboch rodičoch. Genetické ochorenia tohto typu sa vyskytujú s rovnakou frekvenciou u novorodencov chlapcov a dievčat. Patogenéza PKU je determinovaná metabolickou poruchou fenylalanínu, ktorá sa môže vyskytovať v 3 formách. Len klasická fenylketonúria typu I sa môže liečiť diétnou terapiou.
Atypické formy ochorenia nie je možné vyliečiť úpravou diéty. Tieto odchýlky sú spôsobené nedostatkom tetrahydropterínu, dehydropterín reduktázy (menej bežne pyruvoyl tetrahydropterín syntáza, guanozín-5-trifosfát cyklohydrolaza, atď.). Väčšina prípadov smrteľných výsledkov bola zaznamenaná u pacientov so zriedkavými variáciami PKU, zatiaľ čo klinické prejavy všetkých foriem ochorenia sú podobné. Riziko výskytu dieťaťa s mutovaným génom fenylalanín-hydroxylázy sa zvyšuje, ak sú jeho rodičia blízki príbuzní (v príbuzných manželstvách).
diagnostika
Ak je podozrenie na genetickú poruchu, diagnóza sa stanoví na základe kombinácie údajov získaných zo štúdia histórie ochorenia - genealogických informácií, výsledkov klinického a lekárskeho genetického výskumu. Na včasné odhalenie vrodených chorôb (PKU, cystická fibróza, galaktozémia atď.) Bol vyvinutý povinný skríningový program pre všetky novorodencov (neonatálny skríning).
Ak budúci rodičia vedia o preprave mutovaného génu, moderná medicína ponúka spôsoby na odhalenie defektu počas tehotenstva (prenatálna diagnostika plodu invazívnou metódou). Na separáciu fenylketonúrie na druhy podľa závažnosti sa používa podmienená klasifikácia, ktorá je založená na hladine fenylalanínu v tekutine bez fibrínu získanej z krvnej plazmy:
- Ťažká fenylketonúria - 1200 µmol / l.
- Priemer je 60-1200 μmol / l.
- Svetlo (nevyžaduje liečbu) - 480 µmol / l.
Test skríningu
Identifikácia genetických abnormalít sa vyskytuje v niekoľkých štádiách. V prvej fáze pôrodnice, pre všetky deti vo veku 3-5 dní, sa odoberá periférna krv (z päty) na výskum. Materiál sa aplikuje na papierovú formu a zasiela do biochemického laboratória, kde sa vykonáva biochemická analýza. V druhej fáze skríningového testu sa zistí, že koncentrácia fenylalanínu je normálna.
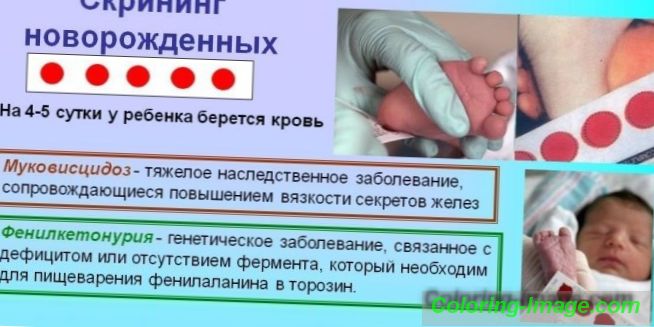
Ak sa nezistia žiadne patologické zmeny, diagnostika sa dokončí a vykoná sa zápis na detskej karte. V prítomnosti odchýlok od normy sa výsledky diagnózy zasielajú pediaterovi, aby poskytli objasňujúce vyšetrenie vzorky krvi novorodenca. Zdravie dieťaťa závisí od včasného a presného vykonania všetkých opatrení na identifikáciu odchýlok. Ak sa diagnóza potvrdí po opätovnom skríningu, rodičia dieťaťa sa odporučia na kliniku detskej genetiky na účely liečby.
Analýzy a štúdie na potvrdenie diagnózy
Opakovaná diagnostika, keď sa počas počiatočného skríningového testu zistia abnormality, sa vykonáva opakovanými testami. Okrem stanovenia obsahu fenylalanínu v krvi, metódy diagnostikovania PKU u detí a dospelých zahŕňajú: \ t
- Test kácenia - stanovenie kyseliny fenylpyrohroznovej v moči pridaním chloridu železitého do biomateriálu (dôjde k modrozelenému zafarbeniu);
- Guthrieho test - posúdenie stupňa reakcie mikroorganizmov na metabolické produkty alebo enzýmy obsiahnuté v krvi pacienta;
- chromatografia - štúdium chemických vlastností látok rozdelených medzi dve fázy;
- fluorimetria - žiarenie biomateriálu monochromatickým žiarením na určenie koncentrácie látok v ňom obsiahnutých;
- elektroencefalografia - diagnostika elektrickej aktivity mozgu;
- Zobrazovanie magnetickou rezonanciou je excitáciou atómových jadier buniek elektromagnetickými vlnami a meraním ich odozvy.
Liečba klasickej fenylketonúrie
Základom terapie fenylketonúrie je obmedzenie konzumácie produktov, ktoré sú zdrojom živočíšnych a rastlinných proteínov. Jedinou metódou úspešnej liečby je diétna terapia, ktorej primeranosť je hodnotená obsahom fenylalanínu v krvnom sére. Maximálna povolená hladina aminokyselín u pacientov rôznych vekových skupín je:
- u novorodencov a detí do 3 rokov - do 242 µmol / l;
- u detí predškolského veku - do 360 µmol / l;
- u pacientov vo veku od 7 do 14 rokov - do 480 µmol / l;
- u adolescentov až do 600 μmol / l.
Účinnosť diéty závisí od štádia, v ktorom bola choroba upravená. S včasnou diagnózou vrodenej patológie sa diétna terapia predpisuje od 8. týždňa života (po tomto období začínajú nezvratné zmeny). Nedostatok včasných opatrení vedie k komplikáciám a zníženiu úrovne inteligencie o 4 body na 1 mesiac od momentu narodenia do začiatku liečby.

Vzhľadom na to, že terapeutická strava pre fenylketonúriu znamená úplnú elimináciu živočíšnych bielkovín z potravy, je potrebné použiť iné zdroje esenciálnych aminokyselín, ako aj vitamíny skupiny B, minerálne zlúčeniny obsahujúce vápnik a fosfor. Produkty označené ako doplnky stravy bez obsahu bielkovín zahŕňajú: \ t
- proteínové hydrolyzáty (Amigen, Aminazol, Fibrinosol);
- zmes bez fenylalanínu nasýtená esenciálnymi aminokyselinami - tetrafen, bez fenylu.
Spolu s nápravnými opatreniami na odstránenie príčiny zhoršeného fungovania organizmu by sa mala vykonať symptomatická liečba zameraná na elimináciu defektov reči a normalizáciu koordinácie pohybov. Kombinovaná terapia zahŕňa fyzioterapeutické procedúry, masáže, pomoc logopéda, psychológa, gymnastické cvičenia. V niektorých prípadoch je spolu s diétnou terapiou indikované použitie antikonvulzív, nootropných a vaskulárnych liekov.
Vlastnosti liečby atypických foriem
Fenylketonúria typu II a typu III sa nedá liečiť diétou s nízkym obsahom bielkovín - hladina fenylalanínu v krvi zostáva nezmenená, keď je príjem bielkovín v tele obmedzený alebo sa klinické príznaky vyvíjajú aj pri poklese hladiny aminokyselín. Účinná liečba týchto foriem ochorenia sa uskutočňuje pomocou:
- tetrahydrobiopterínový faktor postihnutého enzýmu;
- syntetické analógy tetrahydrobiopterínu - tieto látky lepšie prenikajú cez hematoencefalickú bariéru;
- lieky na substitučnú liečbu - nevylučujú príčinu fenylketonúrie, ale podporujú normálne fungovanie tela (Levodopa spolu s karbidofou, 5-hydroxytryptofánom, 5-formyl tetrahydrofolátom);
- hepatoprotektory - podporujú fungovanie pečene;
- antikonvulzíva;
- zavedenie génu fenylalanín hydroxylázy v pečeni - experimentálna metóda.
Zvláštnosti výživy novorodencov a diétna terapia
V prvom roku života dieťaťa s PKU je dojčenie prijateľné, ale jeho množstvo by malo byť obmedzené. Do 6 mesiacov je prijateľná hladina príjmu fenylalanínu 60-90 mg na 1 kg hmotnosti dieťaťa (100 g mlieka obsahuje 5, 6 mg fenylalanínu). Od 3 mesiacov by sa mala diéta dieťaťa postupne rozširovať o ovocné šťavy a zemiakovú kašu.
Deti od 6 mesiacov umožnili zavedenie do zeleninového pyré, kaše (zo sága), bezproteínových bozkov. Po 7 mesiacoch, môžete dať svoje dieťa s nízkym obsahom bielkovín cestoviny, od 8 mesiacov - chlieb, ktorý neobsahuje bielkoviny. Vek, do ktorého je potrebné obmedziť príjem bielkovín v tele chorého dieťaťa, nebol stanovený. Lekári doteraz diskutujú o realizovateľnosti celoživotnej diétnej terapie, ale súhlasia s tým, že je potrebné dodržiavať diétnu diétu najmenej 18 rokov.
Fenylketonúria, diagnostikovaná u ženy, nie je dôvodom na odmietnutie porodiť dieťa. Nastávajúce matky s PKU, aby sa zabránilo poškodeniu plodu počas tehotenstva a prevencia možných komplikácií, sú potrebné pred plánovaným počatím a pri prenášaní dieťaťa na sledovanie diéty s obmedzením fenylalanínu (jeho hladina v krvi by mala byť až 242 µmol / l).
Prípravok bez laktózy pre deti
Diéta s fenylketonúriou je založená na významnom znížení dávky prírodného proteínu v dennej strave, ale telo novorodenca sa nemôže vyvinúť normálne v neprítomnosti potrebných mikroživín. Na uspokojenie potrieb dieťaťa v proteíne sa používajú zmesi aminokyselín neobsahujúce laktózu, ktoré by podľa ruskej legislatívy mali byť pacientom poskytované bezplatne.
Tolerancia dojčiat na fenylalanín počas prvého roka života sa rýchlo mení, preto je potrebné monitorovať jeho koncentráciu v krvi dieťaťa a vykonať úpravy stravy. Zmesi sú určené pre špecifické vekové skupiny:
- deti do jedného roka sú menované Afenilak 15, Analog-SP, PKU-1, PKU-mix, PKU Anamix;
- deti staršie ako 1 rok majú predpísané zmesi obohatené o vitamíny a minerály s vysokým obsahom bielkovín - PKU Prima, P-AM Universal, PKU-1, PKU-2, XP Maxameyd, XP Maxamum.

Diétne výrobky na doplnenie proteínových rezerv
Jednou z hlavných zložiek potravinovej diéty s fenylketonúriou sú potraviny s nízkym obsahom bielkovín na báze škrobu. Tieto doplnky obsahujú kazeínový hydrolyzát, tryptofán, tyrozín, metionín, dusík a poskytujú dennú potrebu dieťaťa, čo je nevyhnutné pre normálny vývoj a rast. Špecializované produkty, ktoré kompenzujú nedostatok esenciálnych minerálov a aminokyselín, keď im chýba diéta, sú:
- Berlofen;
- Tsimorgan;
- Minafen;
- Aponte.
Strava pre predškolské deti a školákov
Ako sa telo prispôsobuje fenylalanínu, deti od 5 rokov môžu postupne znižovať diétne obmedzenia. Расширение рациона происходит путем введения круп, молочных продуктов, мясных изделий. Школьники старших классов имеют уже высокую толерантность к фенилаланину, поэтому в этом возрасте можно продолжить расширение диеты, при этом необходимо отслеживать реакцию на все изменения в питании. Для контроля состояния ребенка применяются следующие способы:
- оценка неврологических показателей, психологического состояния;
- контроль показателей электроэнцефалограммы;
- определение уровня фенилаланина.
Группы продуктов при ФКУ
В рацион питания пациентов с ФКУ наряду с малобелковыми крахмалистыми продуктами и лечебными смесями входят и продукты натурального происхождения. При составлении меню следует четко рассчитывать количество потребляемого белка и не превышать рекомендованную врачом дозировку. Для исключения токсического влияния на организм разработаны 3 списка продуктов, которые содержат запрещенные (красный), нерекомендованные (оранжевый) и разрешенные (зеленый) позиции.
Красный список
Фенилкетонурия развивается на фоне отсутствия фермента, превращающего фенилаланин в тирозин, поэтому высокое содержание белка является поводом для отнесения продуктов в запрещенный (красный) список. Позиции из этого перечня следует полностью исключить рациона питания больного ФКУ:
- mäso;
- внутренние органы животных, субпродукты;
- klobásy, klobásy;
- морепродукты (в т.ч. рыба);
- яйца всех птиц;
- fermentované mliečne výrobky;
- orechy;
- плоды бобовых и зерновых культур;
- sójové výrobky;
- желатинсодержащие блюда;
- cukrovinky;
- аспартам.
Оранжевый список
Продукты, которые должны дозировано поступать в организм ребенка с диагнозом ФКУ, включены в оранжевый список. Включение в рацион питания позиций из этого перечня допустимо, но в строго ограниченном количестве. Эти продукты хоть и содержат не много белка, но тоже могу повысить уровень фенилаланина, поэтому их употребление не рекомендовано:
- Konzervovaná zelenina;
- блюда из картофеля и риса;
- kapusta;
- mlieko;
- щербет.
Зеленый список
Безбелковые продукты разрешены к употреблению больными с диагнозом фенилкетонурия без ограничений. Перед покупкой позиций из зеленого списка необходимо изучить состав, указанный на упаковке, и убедиться, что там не содержится краситель аспартам, содержащий фенилаланин:
- ovocie;
- овощи (за исключением картофеля и капусты);
- jahody;
- greeny;
- крахмалистые крупы (саго);
- мед, сахар, варенье;
- мучные изделия из кукурузной или рисовой муки;
- масла, жиры (сливочное, растительное, оливковое).
Как контролировать уровень фенилаланина в крови
Фенилкетонурия является неизлечимым заболеванием, которое можно перевести в фазу стагнации путем применения диетотерапии и лечебно-профилактических мер. При изменении условий жизни, нарушении режима питания болезнь может опять обостриться, поэтому больным требуется пожизненное наблюдение. Процесс контроля заключается в периодическом определении уровня фенилаланина в крови. Частота сдачи анализов зависит от возраста пациента:
- до 3 месяцев – скрининг крови надо делать еженедельно до получения устойчивых результатов;
- от 3 месяцев до 1 года – 1-2 раза в месяц;
- от 1 до 3 лет – 1 раз за 2 месяца;
- старше 3 лет – ежеквартально.

Кровь для анализов сдается через 3-4 часа после приема пищи. Помимо скрининга развитие ФКУ контролируется путем определения нутритивного статуса, физического, эмоционального развития больного, уровня интеллектуальных способностей и развитости речи. По результатам наблюдений может возникнуть необходимость дополнительной диагностики с привлечением соответствующих специалистов.